金币
UID176556
帖子
主题
积分25051
注册时间2014-6-6
最后登录1970-1-1
听众
性别保密
|
欢迎您注册蒲公英
您需要 登录 才可以下载或查看,没有帐号?立即注册
x
An Analysis Of FDA Warning Letters On Data Governance & Data Integrity

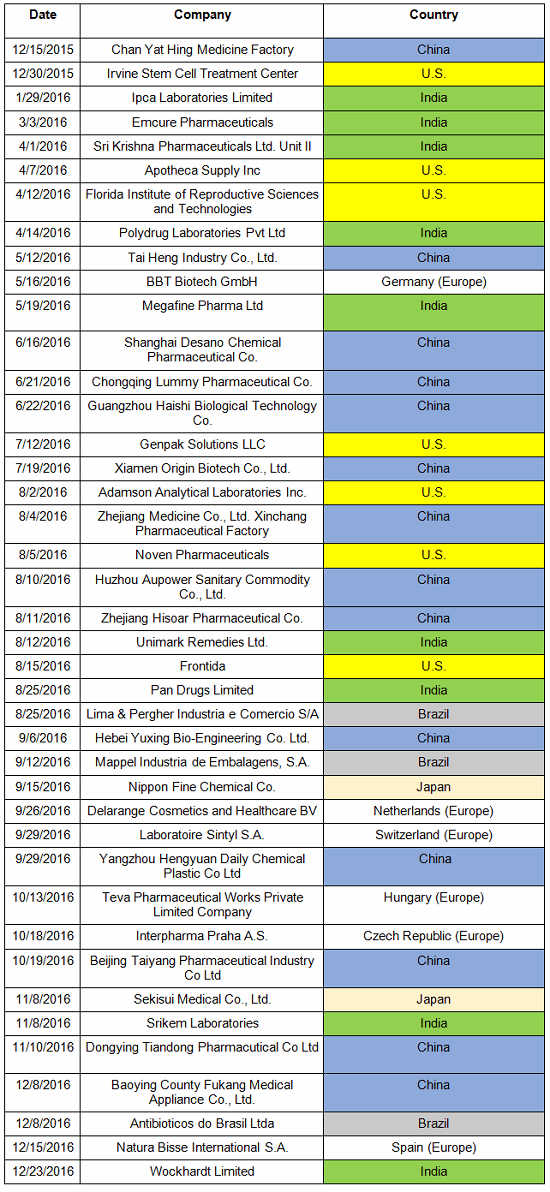
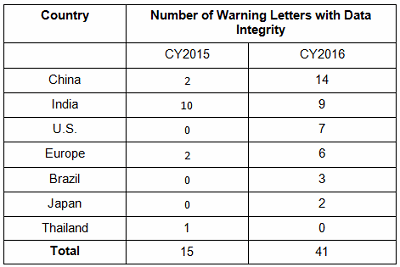
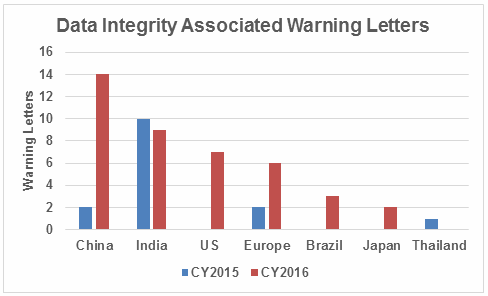
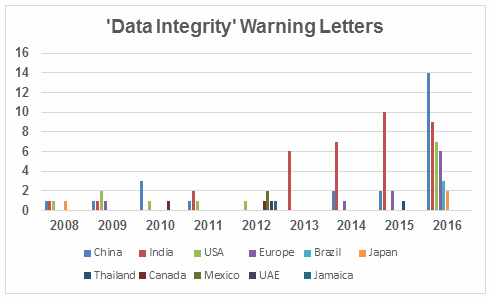
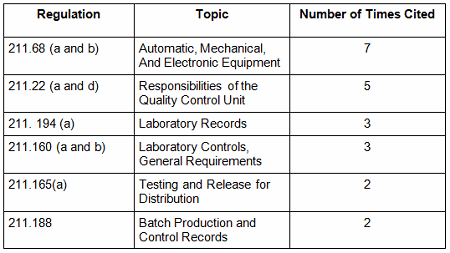
Failures in data governance and data integrity have been more prominent in health authority enforcement actions in the past few years. Health authorities review thousands of pages of data in original submissions and supplements covering the broad GxP area. Inspectors then review manufacturing and test data on-site during pre-approval inspections and routine GMP inspections. However, they cannot even begin to review all the data that is generated, so they must trust that firms are accurately recording and retaining all data and addressing problems appropriately when they arise. This trust is broken when health authorities identify issues such as falsification of records, destruction of original records within their retention period, and practices that demonstrate a lack of control over both paper and electronic documentation. When this happens, firms often suffer serious consequences including warning letters, import alerts, recalls, and consent decree agreements. More than 15 years ago the FDA issued warning letters that identified failures in computer system validation, failure to review laboratory computer systems audit trails, failure to maintain electronic records and failure to investigate their disappearance. While the FDA led the way in early enforcement, global health authorities have now begun to catch up and cite similar deficiencies in their inspections. Approximately 80 percent of all warning letters in 2015 and 2016 include a data integrity component, and approximately 70 percent of the published Eudra reports of GMP non-compliance cite similar shortcomings. In 2015, we published the collection of FDA GMP warning letters that included deficiencies in data governance and data integrity. Here we provide the same information for 2016 drug GMP warning letters. These should serve as a resource for GMP audit staff and QA staff evaluating their own firms and contract sites for gaps in these areas. Table 1 lists the warning letters that include data integrity deficiencies, the date of issuance, and the country where the facility is located. Note the first two warning letters were from inspections in 2015, but published in 2016 and were not counted in last year’s tally. I’ve color-coded the country column and included all European countries in a single group. Table 1: Warning Letters with Data Integrity Deficiencies, 2016 Table 2 identifies the warning letters per country for this year’s tally and for last year’s. This year, China received the most warning letters of this type, with India close behind. Note that in CY2016, seven firms in the U.S. received warning letters with data integrity deficiencies, up from zero the previous year. Brazil and Japan are new to the list this year with three and two warning letters, respectively. Figure 1 shows the same information as a graphic. Table 2: Number of Data Integrity Associated Warning Letters by Country Figure 1: Data integrity warning letters by country, CY2015 and CY2016 Figure 2 captures data from 2008 through 2016 on a country-by-country basis. Some countries are present in multiple years, including the U.S., China, India, and the “Europe” category. A few countries are present only in one or two years. Note the U.S. had warning letters with data integrity deficiencies in all years except 2013, 2014, and 2015 and is the third most frequent country for 2016 warning letters of this type. Figure 2: Data integrity warning letters by country, CY2008 through CY2016 Table 3 shows the regulations cited in the warning letters. Many of the deficiencies identified in the collection for CY2016 did not cite a governing regulation. Many were “conclusions” or “data integrity remediation” instructions from the FDA to which the firm must respond. Many of the letters were issued to API manufacturers and did not cite 21 CFR 211, which applies to finished product. In addition to the regulations cited below, the following regulations were each cited once: 211.100(b), 1271.50(a), 211.165(e), 211.180(e), 211.137(a), 211.180(a), 211.101, 211.186(a), and 211.42(c). As in the past 15-plus years, the FDA has focused on enforcement of the predicate rules. Table 3: Most Frequent Regulation Citations in CY2016 12 Actions Firms Can Take To Avoid Data Integrity Problems 21 CFR 11, Electronic Records and Electronic Signatures, is 20 years old, yet we continue to see ever-increasing enforcement actions for data integrity in electronic records. The last two years have seen data integrity cited in approximately 80 percent of warning letters and increasing participation in this area by other regulatory authorities. The U.K.’s Medicines and Healthcare Products Regulatory Agency (MHRA) was the earliest to enter the area in 2015 with their guidance and a published draft revision in 2016. European Medicines Agency (EMA), World Health Organization (WHO), Pharmaceutical Inspection Co-operation Scheme (PIC/S), Australia, Canada, and China followed in 2016. Further, this is not limited to the GMP area, but now includes GCP, with the most dramatic cases where these problems are uncovered at sites that perform bioavailability and bioequivalence studies. For these firms, hundreds of products are impacted, and sponsors must consider whether data is trustworthy and frequently must perform the studies again at a different site. So, how can a firm prevent, detect, and remediate these problems before the regulators become involved? Here’s the 12-step program: General
- Develop and sustain a corporate culture where reporting mistakes is encouraged without retaliation.
- Trust that this initiative on the regulators’ part is here to stay. It is essential that regulatory authorities can trust the data firms generate.
- Read and understand the intent and application of relevant regulations and guidance. The WHO guidance and the MHRA guidance give discrete examples that are relevant for most authorities.
- Monitor enforcement actions including form 483s, warning letters, import alerts, EU reports of GMP non-compliance, and WHO notices of concern. All of these, except for the form 483s, are available without cost on the Internet, and form 483s can be purchased from commercial sources. Most health authorities give presentations at industry trade group meetings, and their slide decks provide additional insight. Implement what you learn in your internal and external GxP audit programs.
- Remember data integrity applies to paper records and also where paper records and electronic records may intersect.
- Don’t relegate compliance of GxP computerized systems to the IT department alone. They have the technical expertise in this area but are likely not experts in GMP requirements. This is a partnership with many stakeholders between both IT and quality leadership.
Specific
- Develop and implement a data governance process where GxP data is considered a valuable corporate asset that must be accurate, trustworthy and secured through its life cycle.
- Map data and process flows for all computer systems including enterprise systems with GMP components, laboratory systems, and manufacturing systems. This information is key to identifying and implementing risk-based actions.
- Validate systems for their intended purpose taking a risk-based approach. Systems include computer hardware, software, peripheral devices, network infrastructure, operators, and associated documents including standard operating procedures. It is not sufficient to purchase “Part 11 compliant” software because it does not address these other components.
- Assess the gaps in the systems mentioned in 8 above, taking into consideration the predicate rule requirements in addition to the specific data governance/integrity guidance from health authorities. Remember to address the fundamentals, identify what constitutes original data, and ensure original data and its critical metadata (audit trails) are reviewed.
- Implement interim controls where gaps are identified, document and justify their adequacy, and work toward implementation of fully compliant solutions over a defined timeline.
- Understand that remediation may be costly and time consuming. Additional problems will likely be uncovered along the way. Don’t expect remediation to happen immediately; it’s often a multi-year process. Stick with it.

Make sure your data integrity program is compliant. Develop a strategy after getting the information you need in this on-demand webinar. NEW FDA Data Integrity Draft Guidance – Key Points to Understand
Conclusion Failures in data governance and data integrity continue to be addressed in approximately 80 percent of FDA warning letters issued to both domestic and foreign sites. Requirements in this area apply to both paper records and electronic records. Warning letters continue to cite similar deficiencies to those identified between 1999 and 2006, including computer systems not validated for their intended purpose, lack of controls over computerized systems to prevent access by unauthorized users, and failure to evaluate all original data (including audit trails) generated in testing and to consider the results as part of the lot release decisions. The few new deficiencies identified in the past few years include:
- the use of test injections using actual samples to see whether they will pass when tested as part of a complete sample set, and then ignoring the out-of-specification (OOS) result if they fail
- switching audit trails in chromatography data systems off and then on again to obscure modification or deletion of data
- manipulation of date/time stamps to make it appear samples were tested on a different day than they actually were.
The new deficiencies are identified by FDA investigators who have received detailed training in this area and better understand GxP computerized systems and their capabilities. Data integrity and data governance are no longer an esoteric topic owned by the IT department, as they may have been when Part 11 was published. It is owned by every person in the firm who develops or completes an official GxP record. Firms that ignore this area do so at their peril, both professional and financial. Many publications have reported hundreds of millions of dollars spent on remediation and lost sales and opportunities. It is time to become serious about this area of compliance and incorporate it formally into the quality system. Health authority regulations and guidances provide clear expectations for this area and are widely available. Enforcement actions are publicly available and are superb tools for understanding expectations and for use in education and training of staff. Rx-360, an international pharmaceutical supply chain consortium focused on supply chain security, has developed and published a data integrity library that includes global regulations and guidances, slide presentations given by regulatory authorities, and a large collection of articles on the many aspects of data governance and data integrity. Readers are encouraged to avail themselves of this resource along with the enforcement actions published by the health authorities. Most of the information firms need to know about the topic can be found in these two sources. Note: Readers who want a complete listing of the warning letter deficiencies on this topic can find them on my website for 2015 and 2016 (stating on page 5). About the Author:  Barbara Unger formed Unger Consulting, Inc. in December 2014 to provide GMP auditing and regulatory intelligence services to the pharmaceutical industry, including auditing and remediation in the area of data management and data integrity. Her auditing experience includes leadership of the Amgen corporate GMP audit group for APIs and quality systems. She also developed, implemented, and maintained the GMP regulatory intelligence program for eight years at Amgen. This included surveillance, analysis, and communication of GMP related legislation, regulations, guidance, and industry compliance enforcement trends. Barbara was the first chairperson of the Rx-360 Monitoring and Reporting work group (2009 to 2014) that summarized and published relevant GMP and supply chain related laws, regulations, and guidance. She also served as the chairperson of the Midwest Discussion Group GMP-Intelligence sub-group from 2010 to 2014. Barbara is currently the co-lead of the Rx-360 Data Integrity Working Group. Barbara Unger formed Unger Consulting, Inc. in December 2014 to provide GMP auditing and regulatory intelligence services to the pharmaceutical industry, including auditing and remediation in the area of data management and data integrity. Her auditing experience includes leadership of the Amgen corporate GMP audit group for APIs and quality systems. She also developed, implemented, and maintained the GMP regulatory intelligence program for eight years at Amgen. This included surveillance, analysis, and communication of GMP related legislation, regulations, guidance, and industry compliance enforcement trends. Barbara was the first chairperson of the Rx-360 Monitoring and Reporting work group (2009 to 2014) that summarized and published relevant GMP and supply chain related laws, regulations, and guidance. She also served as the chairperson of the Midwest Discussion Group GMP-Intelligence sub-group from 2010 to 2014. Barbara is currently the co-lead of the Rx-360 Data Integrity Working Group.Before Amgen, Barbara worked for the consulting firm Don Hill and Associates, providing regulatory and quality services to the pharmaceutical industry, and for Eli Lilly and Company in quality and CMC regulatory affairs positions. She began her career in the pharmaceutical / device industry with Hybritech Inc. and received a bachelor's degree in chemistry from the University of Illinois at Urbana-Champaign. You can contact Barbara at bwunger123@gmail.com.
|
|


 |手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 京ICP证150354号 互联网药品信息服务证书编号: (京)-非经营性-2024-0033
|手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 京ICP证150354号 互联网药品信息服务证书编号: (京)-非经营性-2024-0033





 发表于 2017-10-13 16:04:07
发表于 2017-10-13 16:04:07

 置顶卡
置顶卡 变色卡
变色卡
 楼主
楼主 感谢楼主,谢谢分享
感谢楼主,谢谢分享