金币
UID26590
帖子
主题
积分33436
注册时间2012-4-10
最后登录1970-1-1
听众
性别保密
|
欢迎您注册蒲公英
您需要 登录 才可以下载或查看,没有帐号?立即注册
x
[size=113%]Data Reliability Guideline
数据可靠性指南
1. Preface 前言The IPA launched its Quality Forum (QF) in April 2015 to help Indian pharmaceutical manufacturers to achieve parity with global benchmarks in quality. The industry made a commitment to a multi‑year journey to address key issues facing the industry and develop best practices. McKinsey & Co. joined this journey as a knowledge partner.
The QF focused on three priority areas. The first among them was to develop a comprehensive set of Data Reliability Guidelines. It took upon itself the challenge of establishing robust and seamless data management and documentation systems and processes. This initiative found great support from the US FDA, the UK MHRA and the EU EMA. The IPA wishes to acknowledge their contributions and commitment.
The IPA also wishes to acknowledge the CEOs of six member‑companies who have committed their personal time, human resources and provided funding for this initiative.
This Guideline is the outcome of a concerted effort over the last 15 months by senior managers engaged in manufacturing, quality and regulatory functions of six IPA member‑companies. They shared current practices, benchmarked these with the existing regulatory guidance’s from the US FDA and other regulatory bodies such as UK MHRA, WHO, etc., developed a robust draft document and got it vetted by leading subject matter experts and regulatory agencies. The IPA acknowledges their hard work and commitment to quality.
This document, to be released at the IPA’s 2nd Annual Conference 2017 in Mumbai, will be hosted on the IPA website www.ipa‑india.org to make it accessible to all manufacturers in India and abroad.
2. Introduction and Background 介绍和背景Managing data in pharmaceutical industry is challenging, especially when firms are experiencing growth of data volume at an exponential rate. While the industry realizes the magnitude and criticality of these issues, addressing them in time remains a challenge ƒ
Suspicious data quality can result in severe consequences for the organization and ultimately jeopardize the reputation of the organization. All data generated requires close observation and immediate intervention in case of any anomaly. It is imperative that proper checks are put in place to avoid proliferation of bad data in the systems ƒ
Implementing controls and maintaining data without first understanding the regulations and business processes can result in data of questionable authenticity, and may lead to regulatory, civil, or criminal action. Ensuring integrity of critical data and metadata is necessary ƒ
Ensuring data reliability is not only a CGMP requirement; it is also a key enabler of effective management decision‑making. Over a period of time, firms with reliable data systems are likely to benefit from informed decision‑making based on greater transparency and accurate data from the shop‑floor ƒ
Data reliability is fundamental in a pharmaceutica l quality system (PQS) which ensures that medicines taken by consumers are of the required quality. Data reliability requirements apply equally to manual and electronic data ƒ
Data reliability applies across the data lifecycle, that is
— Collection of data (including raw data) — Processing and computation of data — Reporting of data — Retention of data, and — Archival/retrieval and authorized destruction Data reliability is considered to be vital, since data should be complete as well as being accurate, legible, contemporaneous, original and attributable, commonly referred to as “ALCOA” ++ where ‘+’ stands for ‘Complete, Consistent, Enduring and Available.’ A more detailed explanation is given later in this Guideline.
3. Scope 范围This Guideline will be applicable to all functions and departments across an organization where GXP activities generate data through systems and processes, i.e.,
— Manufacturing of finished drug products and drug substance for clinical trials, bioequivalence studies, and commercial distributions — Laboratories that develop methods or formulations intended to support a new drug application or laboratories that analyze samples generated from clinical trials — Contract manufacturing organizations — Contract research organizations — Contract testing laboratories — Pharmacovigilance — Contractors who provide GMP activities which could affect the quality of the drug reaching the patient ƒ It is applicable to data that is generated and stored by manual (paper–based), electronic, or hybrid systems. The practices within this document are intended to be incorporated into organizational data reliability standards and procedures in order to
— Support the requirements set forth in the quality manual and standards — Define organizational CGXP data reliability requirements — Provide organizational data reliability expectations to be incorporated into internal audits, quality audits of suppliers, contract manufacturers, contract laboratories, self‑inspections and risk reviews 4. Purpose 目的The purpose of this Guideline is to
— Describe the requirements of maintaining complete, accurate, truthful and verifiable data in all CGXP documents that are needed to be maintained as per regulatory requirements and various governmental regulations, laws, rules and statutes applicable to an organization in this matter — Describe the importance of data generation, maintaining data lifecycle, data governance and data reliability throughout manufacturing sites of an organization including contactors and service providers — Emphasize the paramount importance of ethics, agreements, and understand the regulatory implications of data falsification and fraud ƒ This Guideline will outline a holistic approach, with different elements necessary to help ensure the reliability of data throughout the product lifecycle. The key elements considered for overall data reliability guidelines are
— Culture — Awareness and capability — Process design — Data reliability risk detection and mitigation — Technology and IT systems — Governance ƒ This Data Reliability Guideline focuses on ensuring quality, safety and efficacy – three attributes that are inseparable for all medicines manufactured
5. Definitions 定义Archiving: 归档 Archiving is the process of protecting records from the possibility of further alteration or deletion, and storing these records under the control of dedicated data management personnel throughout the required records retention period ƒ
Audit Trail: 审计追踪 An audit trail is a process that captures details such as additions, deletions, or alterations of information in a record, either paper or electronic, without obscuring or over‑writing the original record. An audit trail facilitates the reconstruction of the history of such events relating to the record regardless of its media, including the “who, what, when and why” of the action ƒ
Backup: 备份 Backup refers to a true copy of the original data that is maintained securely throughout the records retention period. For example, the backup file shall contain data (including associated metadata) and shall be in the original format or in a format compatible with the original format and shall be maintained for the purpose of disaster recovery. The backup and recovery processes must be validated ƒ
Breach of Data Integrity (BDI): 违反数据可靠性 It is a violation of the integrity of data. This means that the actions performed and the documents/records written do not reflect the truth and the reality which has taken place. Breaches of integrity can be observed during manufacturing and testing, inspection and post inspection ƒ
Computerized System: 计算机化系统 Computerized System collectively controls the performance of one or more automated business processes. It includes computer hardware, software, peripheral devices, networks, personnel and documentation, e.g. manuals and standard operating procedures ƒ
Data: 数据 Data means all original and master records and certified true copies of original records, including source data and metadata and all subsequent transformations and reports of this data, which are recorded at the time of the GXP activity, and allow full and complete reconstruction and evaluation of the GXP activity ƒ
Data Governance: 数据管理 This refers to the sum total of arrangements which ensure that data, irrespective of the format in which it is generated, are recorded, processed, retained and used in order that a complete, consistent and accurate record throughout the data lifecycle is maintained ƒ
Data Integrity Assurance: 数据可靠性保障 Assuring data integrity requires appropriate quality and risk management systems, including adherence to sound scientific principles and good documentation practices. Data Integrity requires adherence to the criteria of ALCOA+ as mentioned earlier in this Guideline. The specific definitions are as below
| Criterion | Meaning | | Attributable | Attributable means information is captured in the record so that it is uniquely identified as executed by the originator of the data (e.g. a person, and/or a computer system). | | Legible | The terms legible, traceable,and permanent,refer to the requirements that data are readable, understandable and allow a clear picture of the sequencing of steps or events in the record. | | Contemporaneous | Contemporaneous is the process of documentation (on paper or electronically) at the time of the occurrence of an activity. | | Original | Original data includes the first or source capture of data or information and all subsequent data required to fully reconstruct the conduct of the GXP activity. | | Accurate | Accurate means that data are correct, truthful, valid and reliable. | | Complete | Complete means that all data from an analysis, including any data generated before a problem is observed, data generated after repeating part or all of the work, or re‑analysis performed on the sample are contained the data record. For hybrid systems, the paper output must be linked to the underlying electronic records used to produce it. | | Consistent | Consistent means that all elements of the analysis, such as the sequence of events, follow on and data files are date (all processes) and time (when using a hybrid or electronic systems) stamped in the expected order are contained in the record. | | Enduring | Enduring means that all data have been recorded on authorized media which can be preserved for a period of time, e.g. laboratory notebooks, numbered worksheets, for which there is accountability, or electronic media. Data recorded on scrap paper or any other media which can be discarded later, e.g. backs of envelopes, laboratory coat sleeves or Post‑It notes, etc. are not considered enduring. | | Available | Available means that the complete collection of records can be accessed or retrieved for review and audit or inspection over the lifetime of the record. |
Data Lifecycle: 数据生命周期 This refers to a planned approach to assessing and managing risks to data in a manner commensurate with the potential impact on patient safety, product quality and/or the reliability of the decisions made throughout all phases of the process by which data is created, processed, reviewed, analyzed, reported, transferred, stored and retrieved, and continuously monitored until retired ƒ
Data Owner: 数据所有人 An individual or a team who is responsible for data generation and storage ƒ
Data Reliability (DR): 数据可靠性 Data Reliability is the degree to which a collection of data is complete, consistent and accurate throughout its data lifecycle. The collected data should be attributable, legible, contemporaneously recorded, accurate and should be an original or a true copy ƒ
Data Reliability Auditors (DRAs): 数据可靠性审计人员 Data Reliability Auditors are independent auditors who report to the Corporate Quality (Data Reliability Cell‑Head). The responsibility of these auditors is to conduct an independent reliability audit in order to confirm adherence to established requirements for data reliability in all the quality related processes ƒ
Data Reliability Governance: 数据可靠性管理 It refers to all processes of governing by the Data Reliability Task Force through laws, norms, power or language
Data Reliability Task Force: 数据可靠性专门小组 A ‘Task Force’ is a body which governs data reliability globally at all sites
Dynamic Record: 动态记录 This refers to records in dynamic format, such as electronic records, that allows for an interactive relationship between the user and the record content. For example, electronic records in database formats allow the ability to track, trend and query data; chromatography records, maintained as electronic records, allow the user to reprocess the data, view hidden fields with proper access permissions, and expand the baseline to view the integration more clearly. Examples of dynamic data in quality control are chromatogram, spectrum, LIMS data, PLC and SCADA based data like ‑autoclave printouts, stability chamber monitoring records, KF titrator data, etc. Examples of dynamic data in ware houses would include SAP data base. In manufacturing, dynamic data would include BMS‑Temperature and RH print outs, etc.
Electronic Data: 电子化数据 This includes data from ERP software used for controlling quality systems, laboratory electronic data and records, etc. ƒ
Ethics: 准则 Ethics (also moral philosophy) is the branch of philosophy that involves systematizing, defending, and recommending concepts of right and wrong conduct ƒ
Exception Report: 例外报告 This refers to a validated search tool that identifies and documents predetermined ‘abnormal’ data or actions, which require further attention or investigation by the data reviewer ƒ
GXP: 良好X实践 An acronym for the group of good practice guides governing the preclinical, clinical manufacturing and post‑market activities for regulated pharmaceuticals, biologics, medical devices, such as GLP (good laboratory practices), GCP (good clinical practices), GMP (good manufacturing practices) and GDP (good documentation/distribution practices) ƒ
Hybrid System: 混合系统 A Hybrid System is defined as an environment consisting of both electronic and paper‑ based records (frequently characterized by handwritten signatures executed on paper). A very common example of such a system is one in which the system user generates an electronic record using a computer‑ based system (e‑batch records, analytical instruments, etc.) and then is required to sign that record as per the Predicate Rules (GLP, GMP, GCP, etc.). However, the system does not have an electronic signature option, so the user has to print out the report and sign the paper copy. Now, he/she has an electronic record and a paper/handwritten signature. The system has an electronic and a paper component, hence the term ‘Hybrid System’ ƒ
Metadata: 元数据 Metadata is the data that describes the attributes of other data, and provides context and meaning. Typically, these are data that describe the structure, data elements, inter‑relationships and other characteristics of data. It also permits data to be attributable to an individual. Metadata is structured information that describes, explains, or otherwise makes it easier to retrieve, use, or manage data. (For example, the number “23” is meaningless without metadata, such as an indication of the unit “mg.”). Data should be maintained throughout the record’s retention period with all associated metadata required to reconstruct the CGMP activity. The relationships between data and their metadata should be preserved in a secure and traceable manner. Among other things, metadata for a particular piece of data could include a date/time stamp for when the data was acquired, a user ID of the person who conducted the test or analysis that generated the data, the instrument ID used to acquire the data, audit trails, etc ƒ
Paper-based Data: 基于纸质媒介的数据 This includes recording formats (such as worksheets and logbooks), batch records, master records, green sheets, apex, but are not limited to documents alone ƒ
Quality Risk Management (QRM): 质量风险管理 This refers to a systematic process for the assessment, control, communication and review of risks to the quality of the drug (medicinal) product across the product lifecycle (ICH Q9) ƒ
Raw Data: 原始数据 This refers to original records and documentation, retained in the format in which they were originally generated (i.e. paper or electronic), or as ‘true copies’. Raw data must be contemporaneously and accurately recorded by permanent means. In the case of basic electronic equipment which does not store electronic data, or provides only a printed data output (e.g. balance or pH meter or chart recorder), the printout constitutes the raw data ƒ
Senior Management: 高级管理 This refers to the person(s) who direct and control a company or site at the highest levels with the authority and responsibility to mobilize resources within the company or site (ICH Q10 based in part on ISO 9000:2015) ƒ
Static Record: 固态报告 A static record format, such as a paper or PDF record, is one that is fixed and allows no or very limited interaction between the user and the record content. For example, once they are printed or converted into static PDF files, chromatography records lose the capabilities of being reprocessed or enabling more detailed viewing of baselines or viewing of hidden fields. Examples of static data in quality control include pH melting point results reported in worksheets, or in paper logbooks; in warehouses, material de‑dusting records, goods receipt notes, container physical verification records; in manufacturing, paper‑based BMR in which complete activity of batch manufacturing is recorded, cleaning records of equipment’s and instruments, etc. ƒ
Subject Matter Experts (SMEs): 特定领域专家 A subject‑matter expert (SME) or domain expert is a person who is an authority in a particular area or topic ƒ
True Copy: 真实副本 A true copy is a copy of an original recording of data that has been certified to confirm it is an exact and complete copy that preserves the entire content and meaning of the original record, including in the case of electronic data, all metadata and the original record format as appropriate ƒ
Wrongful Act: 错误行为 Wrongful act means employee conduct that raises significant questions regarding data reliability involving fraud, falsification and untrue statements, misconduct, wrongdoing or other acts that subvert the integrity of data for a regulated product that is required to be maintained in accordance with company policies, standards of procedures, or in accordance with applicable laws, regulations or legislative directive of regulatory authority or authorities
6. Responsibilities 职责Management is responsible for
— Ensuring that this Guideline is implemented across the entire organization in order to ensure that a robust and sustainable data management system and governance is in place — Establishing and maintaining organization‑wide commitment to data reliability as an essential element of the quality system — Ensuring that personnel are not subject to commercial, financial and other pressures or conflicts of interest that may adversely affect data reliability — Making staff aware of the relevance of data reliability and its importance — Creating a work environment in which staff is encouraged to communicate failures and mistakes related to data reliability so that corrective action can be taken — Building systems and controls which prevent employees from altering or falsifying data — Creating channel(s) for employees to report any breaches in data reliability to senior management — Ensuring adequate information flow between staff at all levels — Discouraging any management practices that might be expected to inhibit the active and complete reporting of data reliability related issues — Reviewing quality matrix related to data reliability and key performance indicators of the quality management system — Encouraging employees to take an open‑door approach and to take appropriate actions as per open‑ door privileges for employees — Taking appropriate disciplinary actions on employees for unethical conduct for data reliability. — Site Management shall be responsible for identifying SMEs for each function — Understanding resource constraints that may lead to breach in data reliability and/or integrity and for communicating to staff that such resource constraints should never lead to breach in data integrity ƒ Subject Matter Experts (SMEs) are responsible for
— Performing data process lifecycle mapping, gap assessment, risk identification and mitigation. — Developing data reliability checklists and review processes. — Designing and updating training materials and imparting trainings as required. ƒ Data Reliability Compliance Head is responsible for
— Maintaining overall data governance functionality — Notifying to Global Quality Head and Senior Management about incidence and discrepancies raised during data reliability inspections by Data Reliability Auditors — Identifying root cause for data reliability issues in association with site management and SMEs and providing corrective and preventive action according to regulatory inspections — Providing guidance to the respective SMEs for identifying risk associated with data reliability and proposing suitable corrective and preventive action and monitoring implementation and effectiveness — Providing inputs in overall improvement in quality metrics ƒ Data Reliability Auditors are responsible for
— Performing scheduled and unscheduled data reliability assessments (DRAs) and inspections at sites as per authorized data reliability checklists with the help of trained data reliability auditors — Ensuring compliance related to the discrepancies identified during the inspection ƒ All Employees are responsible for
— Following this Guideline. Responsibilities related to data reliability will be communicated to each level of employee through a code of conduct document as mentioned in Annexure 1 and each employee will acknowledge the same 7. Culture 文化Culture is the foundation for a strong data reliability mindset. Data reliability culture can be improved through training, communication and change management. Common understanding, awareness of impact, ownership and leadership support is required for developing a culture ƒ
This will aid in creating an environment needed to facilitate every individual in guiding his/her own behavior to
— Work in the interest of the organization — Manufacture quality drugs for the patients, and — Improve these behavioral aspects continually ƒ This section focuses on developing a data reliability mindset, a mandate for each level of the employee, developing best practices and the required actions to be taken in case breaches in data integrity are discovered ƒ
Employee responsibilities related to data reliability will be communicated to each level of employee as a code of conduct and every concerned employee will acknowledge the same ƒ
Employees have a duty to engage in appropriate conduct to ensure that all stake holders can trust employee decisions that are based on data and information which are accurate, thoughtful and complete. (Refer to Annexure 1: “Code of conduct for Data Reliability”) ƒ
A culture of compliance is born out of this foundation and it shapes the decisions and actions taken by the employees of an organization. Hence, an organization should have a corporate policy on Ethical Quality Conduct. Each employee should also take the pledge for ethical quality conduct as a commitment to quality (Refer to Annexure 2: “Ethical Quality Conduct”) ƒ
Analysis of employees
— HR will analyze employees on appropriate factors for behavioral aspects related to data reliability during the hiring process and will continue to carry this out throughout the employee’s career in the organization — HR will use benchmark practices and deploy scientific and objective methodology in the selection process of employees to ensure a greater sense of fairness and transparency in this process. The organization will examine employees’ past behavioral records by data mining and will analyze employees’ intentions and attitudes towards data integrity. Based on these intentions and attitudes, in combination with perceived behavioral controls, the organization will predict future behaviors of employees and utilize these predictions in job assignment and human resource decisions — The organization will also monitor and analyze changes in behavior and attitudes of existing employees based on these parameters and utilize the findings in job assignment and human resource decisions ƒ The key behavioral aspects for the absence of reliable data could be the following — Institutional bad habits: Leadership fails to demonstrate the appropriate behavior. An example of a performance measure that drives wrong behavior is a focus on short‑term gains — Management using ‘Rule by Fear’ method with employees (for example, ‘you do what you are told’). This leads to a culture of fear and blame and an inability of employees to challenge the status‑ quo — Poor education: This could lead to bad decisions or inappropriate behavior based on knowing ‘How’ but not ‘Why’ — Poor attitude toward problems: This could lead to a ‘victim’ mindset rather than a learning mindset, in that problems are seen as “bad” rather than “opportunities to improve” — Complex systems and systems with inappropriate design: These can encourage and, at times, even force bad practices — A hierarchy which does not enable employees: A constructive, enabling hierarchy is needed to provide employees with the knowledge and confidence to make the correct decisions — Panic, stress and fatigue: This can lead to negative behavior like fight, flight or freeze — Lack of personnel integrity and honesty: These are exemplified by attitudes of “don’t care” and “I won’t get caught”. Such attitudes are displayed by employees with very little pride in what they do ƒ These key behaviors are indicators or triggers for BDI, and should be assessed by the organization throughout the hiring process as well as employee engagement and employment during his or her career in the organization ƒ Open-door to Management: An employee should be encouraged to take advantage of an open‑door route to organization top management when it comes to raising compliance issues and discussing potential compliance concerns pertaining to data reliability ƒ
Whistleblower policy: In order to create enduring value for all stakeholders and ensure the highest level of honesty, integrity and ethical behavior in all its operations, the Company should formulate a Whistleblower Policy in addition to the existing Code of Conduct that governs the actions of the employees
— This whistleblower policy shall aspire to encourage all employees to report suspected or actual occurrence(s) of illegal, unethical or inappropriate events (behaviors or practices) that affect the Company’s interest and image due to data reliability issues — Management shall be available to respond to questions and concerns if an employee does not feel comfortable talking with his/her supervisor; they also may directly contact the Senior Management of the company through a helpline for concerns related to data reliability. Reporting to the helpline may be made anonymously. Management shall take appropriate action as required upon receiving the information through the helpline. The helpline number will be easily accessible to each employee ƒ Reporting of data integrity failures to regulatory bodies
— When issues relating to data validity and reliability are discovered, it is important that their potential impact on patient safety and product quality, and on the reliability of the information used for decision‑making and applications are examined as matters of top priority — Respective health authorities shall be notified if the investigation identifies material impact on patients, products, reported information or on application dossiers — Individuals who observe data integrity issues can also report suspected issues of this nature that may affect the safety, identity, strength, quality, or purity of drug products to respective drug regulatory authorities or combination of regulatory agencies and the words “CGMP data integrity” shall be included in the subject line — For product marketed in India, CDSCO/DCGI shall be notified for any failures and breaches observed in data integrity — For USFDA, where it is the reporting authority for such matters, notifications should be sent to Druginfo@fda.hhs.gov — For Europe and other agencies, the respective qualified person will be notified for further communication with agencies ƒ Disciplinary actions: Disciplinary action shall be taken against employees who are found to be responsible for unethical conduct related to data reliability requirements
— Impact on quality due to the unethical conduct of the employee shall be assessed through the applicable quality management system procedure at the site. However, as a part of corrective action, employee will be warned and prevented from performing GXP activities — The organization shall formulate and publish clear disciplinary practices to address situations where an employee is found engaged in illegal or unethical conduct related to data reliability. While misconduct is evaluated on a case‑by‑case basis, the organization will take corrective actions in a consistent manner so as to ensure that such action is appropriate under the circumstances and has the intended deterrent effect. Penalties for compliance violation may include termination of the employee at the sole discretion of the organization — The organization shall take every possible measure to prevent and correct issues that can lead to breaches in data reliability. While evaluating data reliability issues, the organization will look into the root cause of the systemic dysfunction rather than individual misconduct. During inspection by regulatory authorities and in the course of internal review, if a breach in data reliability is discovered, and/or misconduct is observed, then the concerned individual shall be removed from performing GXP operations ƒ Town Hall Meeting:
— The Management shall proactively create global awareness at all the manufacturing sites about importance of data, through actions such as conducting town hall meetings. Such meetings shall be conducted when a serious situation arises and Management wants to convey a message on the organization’s stand on data reliability — Subject matters of communication during town hall meetings shall be such as mentioned below, but need not be limited to these » The organization is committed to “doing the right thing when even no one is watching.” » The behavior of employees must reflect their commitment to work in the interest of the organization and manufacture quality drugs for patients and to continually improve the ability of the organization to do so » Data reliability is important because it ensures the safety, efficacy and assurance of the quality of the drugs that consumers will use, and also because it helps to strengthen the trust that regulatory bodies place on the organization » Non‑reliable data lead to recalls, warning letters, importing alerts, injunctions and/or seizures, as well as decrees from regulatory bodies » Intentional acts by employees that do not support data reliability are subject to disciplinary actions ƒ The organization shall recognize and reward employees for their contributions towards creating and developing a sustained data reliability culture. The evaluation criteria for reward and recognition should be
— Consistent adherence to data reliability guidelines — No cases of breaches in data integrity in a particular month — Efficient and effective efforts of the employee related to the execution of DR guideline 8. Awareness and Capabilities 意识与能力Data reliability might be compromised if employees are not aware of the requirements related to data reliability for GMP activities and/or Quality‑related processes that are performed by them. The individual’s capabilities will be improved through training and awareness ƒ
The organizational management at all levels will ensure that personnel under their responsibility, including contractors and consultants, have the appropriate qualification, experience and training required in assuring data reliability awareness ƒ
Employees will be made aware of the specific requirements related to data reliability for the activities to be performed by them; they will also be trained to maintain current awareness of applicable laws, regulations and legislative directives that pertain to documentation and record keeping. Such trainings will be imparted on a regular basis through mandatory and refresher training, based on his/her job profile, i.e. Operator, Supervisor, or Manager ƒ
Available GMP trend/regulatory landscape, for example, Monitoring Warning Letters, 483’s, WHO‑ NOC, Health Canada Inspection Tracker, EUDRAGMP website, etc. across the industry and applicable regulatory guidelines will be taken into consideration for updating the training related documentation on a regular basis ƒ
The organization shall build the requirement for data integrity into Quality Agreements with contractors, and create awareness among staff so they can assist with this endeavor, and report concerns proactively. Quality agreements shall be in place between manufacturers and suppliers and contract manufacturing organizations (CMOs) with specific provisions for ensuring data integrity across the supply chain. This may be achieved by setting out expectations for data governance, and transparent error/deviation reporting by the contract acceptor to the contract giver. There shall also be a requirement to notify the contract giver of any data integrity failures identified at the contract acceptor site ƒ
The employee‑to‑supervisor feedback process, related to data reliability, shall be taken as an opportunity to impart and improve training and awareness modules. Such training would be in the form of quizzes in order to assess the effectiveness of the training ƒ
The company shall establish and maintain an employee Learning Management System which will include the fundamental training requirements pertaining to documentation of GXP activities, including concepts and principles of data reliability, and how employees are to report suspected data reliability issues to company management ƒ
Following aspects shall be covered in data reliability training, but the topics need not be limited to these
— Data reliability and assurance using ALCOA++ criteria — Impact and consequences of data integrity violation — GDP (Good Documentation Practices) in paper‑based and electronic systems — Expectations from a paper‑based system — Expectations from an electronic system, including assuring integrity of electronic records, restricting access, establishing access control, user privileges, review of audit trials, administrative controls and other related matters — Good Chromatographic Practices — Identifying unreliable lab results for analytical process including mobile phase, suitability solutions, sample preparation, integration peaks — Recording of observations during document review and audits — Data governance — Risk management for data reliability — Role‑based training for doers — Training for SMEs — Data review (paper and electronic) 9. Process design 流程设计All the activities related to quality and/or GMP shall be designed so as to support data reliability across the data lifecycle in order to ensure that data is complete and meet the ALCOA criteria ƒ
Design for paper-based system
— Paper formats shall be controlled and accounted manually. Wherever possible, electronic systems shall be implemented to control and account for paper formats. Archival mechanisms shall be established in such a manner that paper records are secured. Wherever possible, paper records shall be archived with help of electronics having full control over protecting data in its original context — Accurate, legible and contemporaneous recording of paper data shall be monitored and verified with little loss of time from when the data was recorded. Risk‑based review mechanisms shall be included in the design ƒ Given below are dos and don’ts for Good Documentation Practices that shall be followed by the organization while doing documentation design
ƒ"Dos"requirement for GDP
— DD/MM/YY and HH:MM formats shall be followed — Pens using indelible (permanent) inks of specified colors shall be used — GXP data shall be recorded directly on approved and authorized formats — Alterations made to handwritten entries shall be made in the following manner » A single line should be marked through the error followed by signature and date » Alterations shall permit reading of original information » Reason/s for the alterations shall be recorded — Modifications, changes and corrections in the master document shall be carried out through Change Control Procedure only. (No handwritten corrections shall be allowed on master documents.) — Design of recording format shall provide sufficient space and shall have provisions to record entries, signatures and record date/time (as applicable) — ‘NA’ shall be used with signature and date — In multiple blank spaces/rows/columns, a diagonal single line across the whole field or space shall be used, ‘NA’ shall be recorded with signature and date, and a brief justification shall be recorded — On draft documents intended for review, the word ‘DRAFT’ shall be used as a watermark, or a stamp with the word ‘DRAFT’ shall be used to mark all pages — Critical significant steps in documents shall be identified and the same shall be required to be checked by a second person while performing the task — Actual observations shall be recorded in the records. However, in certain formats such as a checklist, the recording of ‘Yes’ or ‘No’ shall be used to indicate whether the activity was performed or not — For analysis and in manufacturing, SOP/BMR/STP shall be followed step by step, that is sequentially, and documentation should happen concurrently — MS® Excel sheets used for calculation shall be validated — Signature Specimen Records for short (initial) and full signature shall be maintained, wherein each employee involved in GXP activities shall give a specimen of his/her signature and initials to ensure that the signature is traceable to the correct member of the staff — In cases where the original documents have attachment(s), there shall be clear indications about the number of attachments. Traceability (Parent‑Child Relationship) shall be maintained in chronological order — While handling thermal paper, the procedure of signature, followed by photocopying and attaching the thermal paper with the photocopy shall be followed — All weight slips, printouts, chromatograms, spectrum, etc. shall be signed after completion of the activity. Print‑outs pasted in the record shall have the relevant signatures across the printouts — Documents shall be maintained without any damage — Electronic records are equally important and are given due attention by regulatory agencies ƒ 'Don'ts' for GDP
— Handwritten corrections on Master Documents are not permitted — Use of ditto marks (e.g.____” ____) to fill repetitive entries is not permitted — Use of third brackets ‘{ }’ is not allowed for signature against multiple entries — Use of pencil or any removable and water soluble ink is not permitted — Use of eraser or ink remover or whitener is not permitted — Recording of data on unauthorized documents (e.g. post‑it‑notes, sticky sheets, scrap paper, personal notebooks), glass boards and black/white boards, etc. is not permitted — Overwriting, multiple crossing of the original entry/data and similar practices are not permitted — An entry made by a person on a GMP document but not signed and dated by the person who has made the entry is not accepted as authentic — Pre‑dating or back‑dating entry is strictly not permitted — Destruction and/or deletion of a record, document or report because of any error or mistake are strictly not permitted. Such a record, document or report is still required for reasons of traceability — Deliberately amending or destroying records, documents or reports to hide or falsify data is strictly forbidden. Such actions shall lead to strict disciplinary action ƒControl on blank forms
— Control on blank forms shall be done as part of the good documentation practices of the organization. Blank forms (including, but not limited to, worksheets, laboratory notebooks, and MPCRs) shall be controlled by the quality unit or by another document control method. For example, numbered sets of blank forms may be issued as appropriate and shall be reconciled upon completion of all issued forms. Incomplete or erroneous forms shall be kept as part of the permanent record along with written justification for their replacement ƒ Signature Practices — The same person shall not sign for multiple roles for one activity — Done by » 'Done by' means 'performed by' and the person is the doer » The doer is a person who is responsible for the activity by means of preparation, doing or performance » The ‘done by’ signature identifies the person who actually did the work and documented it. Thus, the ‘done by’ signature is attributable to the person performing the activity or generating the data » The doer is the person who actually performs the activity and shall record or make entry of observations by him/ her followed by signature and date — Checked by » This identifies the person who witnessed the activity being performed » The checker is a person proficient in the task performed and has been trained to perform the activity » The checker verifies that the doer has recorded contemporaneously » The checker has watched and/or witnessed the activity being performed. The checker may also perform the ‘supervisory recording’ (see below) » For the checker to sign, the document must have the signature of the doer. The checker cannot be the only individual to sign the document » All the critical stages/steps shall be checked by another person — Checked and Recorded by: (supervisory recording, recording by scribes) » ‘Checked and recorded by’ shall carry the initials or signature of a person who is checking or supervising the activity and recording the information of the activity performed, and the readings of an operation which are recorded contemporaneously » The ‘Checked and recorded by’ part of the recording process shall be used only if the operator (doer) performing the operation is unable to initial and date immediately, due to working in a confined or restricted space, or to avoid intervention in the process » The use of ‘supervisory recording’ (scribes) to record activity on behalf of another operator shall be considered ‘exceptional’ and should take place only when normal documenting processes can place the product or activity at risk, e.g. in manufacturing steps such as addition of material in batch or documenting line interventions by sterile operators » Each department shall maintain a list of the activities to which the process of “supervisory (scribe) recording” of the documentation applies. This shall be preapproved by QA — Verified by » The ‘verified by’ signature identifies the person who ensured that the work was performed and documented correctly » He or she verifies that sequential steps were performed and the doer has recorded the entries, based on objective evidence, associated records/logs, etc. » The verifier cannot perform the ‘supervisory recording’ » For the verifier to sign a document, it is must to have signature of the doer. The verifier cannot be the only individual to sign the document; e.g. a document signed ‘Line Clearance by QA’ which is signed only by the verifier shall be considered inadmissible — Reviewed by » The ‘reviewed by’ signature identifies the person who ensured that the work was performed and documented correctly (possibly at a later time) » 'Reviewed by'is the initials or signature of the person who reviews the document or record in order to confirm its accuracy and completeness, clarity and legibility, including checking of the calculations if applicable » The reviewer is a person who is responsible for reviewing the documents based on evaluation of supporting data, documents and/or references attached and the checker’s comments — Approved By (or Authorized By) » The ‘Approved by’ signature identifies the person who evaluates the work performed to ensure it was done accurately, completely, and in accordance with procedures and/or documentation practices » ‘Approved by’ indicates approval to proceed to the next stage or process » The approver is a person who is responsible for approving or authorizing the documents based on evaluation of the critical steps, summary, and final conclusion or comments by the reviewer » The approver may also review the documents for clarity, understanding and decision making — Helper or Assistant » The ‘helper’ provides assistance to the ‘doer’ in order to make it easier for the latter to perform physical activities » Helpers are persons who perform motor activities and help the doers and the supervisors in physical handing, such as moving of trolleys, lifting of bags and containers, etc. Since the helper is only performing physical activities, in the sense of definition, the officer or the person getting such activities done through instructions will be the ‘doer’ — Designee » The designee is a person who is allocated signature and decision‑making authority in absence of his/her superior as per the Deputation Matrix of the organization » The deputation matrix shall be prepared by site and support functions » The allocation of designee is based on his/her knowledge, experience and competency; the designee shall be from the same function ƒ Design of electronic system
— All computerized systems used by organizations shall be evaluated, controlled and managed in accordance with GMP and GDP requirements — To assure the integrity of electronic data, computerized systems will be validated at a level appropriate for their use and application. Validation will address the necessary controls to ensure the integrity of the data, including original electronic data and any printouts or PDF reports — The depth and scope of validation will depends on the diversity, complexity, and criticality of the computerized application — Systems shall be assessed to identify data integrity risks and/or vulnerabilities to manipulation — All computerized systems that have the potential for impact on product quality shall be designed and managed to ensure protection from accidental or deliberate manipulation, modification or any other activity that may impact data integrity — Users will be adequately involved in validation activities to define critical data and data lifecycle controls that assure data integrity ƒ Qualification and Validation of Computerized Systems
— Risk assessments shall be in place for each system emphasizing the required controls to ensure data integrity — Validation shall be performed for each system and a report shall be in place stating at least the following items » Critical system configuration and controls for restricted access to configuration and any changes made therein » A list of currently approved users » Privileges for each user of the system » Identity and role of the System Administrator » Frequency of review of audit trails and system logs » Procedures for new system user creation, deleting users, changing of privileges, backing up, recovery and archiving » Original data shall be retained with relevant metadata in formats that allow the reconstruction of process — Companies should have a Validation Master Plan in place — Systems shall be challenged with defined tests before their routine use to ensure they conform to acceptance criteria — Specific tests in DQ, IQ, OQ and PQ shall be included to challenge data integrity risk areas during qualification testing — Computerized systems shall be evaluated periodically, at a pre‑determined frequency, based on risk assessment depending upon criticality, in order to ensure that they remain in validated state. This evaluation shall include deviation, changes, upgrade history, performance and maintenance — Interfaces shall be designed and assessed in such a way that it allows complete and accurate data transfer between systems ƒ Records
— Static Records » The expectations from paper records include controls for retention of original paper records or certified true copies of original paper records, but are not limited to, static format records » The records shall also include written procedures, training, review and audit, and self‑inspection of processes defining conversion, as needed, of original paper records to true copies, which shall be done in the following steps: copies are made of the original paper records, preserving the original record format, the static format, as required (e.g. photocopy, PDF files, etc.) — Dynamic Records » Expectations from electronic records These shall include written procedures, training, review and audit and self‑inspection of processes defining conversion, as needed, of original electronic records to true copies which shall be done in the following steps 1. Copies are made of the original electronic data set, preserving the original record format, the dynamic format, as required (e.g. backup copy of the entire set of electronic data and metadata using a validated backup process) 2. A second person verifier or a technical verification process (such as use of a technical hash function) shall confirm successful backing up, whereby a comparison is made of the electronic backup copy to the original electronic data set to confirm that the copy preserves the entire content and meaning of the original record (i.e. all of the data and metadata are included, no data is missing in the copy, dynamic record format is preserved as important for record meaning, and the file was not corrupted during the execution of the validated backup process) » Preserving the original electronic data in electronic form is also important since data in dynamic format facilitates greater usability of the data for subsequent processes. For example, temperature logger data maintained electronically facilitates subsequent tracking and trending and monitoring of temperatures in statistical process control charts » There are a few special risk management considerations for retention of original records and/or certified true copies. Certified true copies of electronic records shall preserve the dynamic format of the original electronic data as essential to preserving the meaning of the original electronic data. For example, the original dynamic electronic spectral files created by instruments such as FT‑IR, UV/Vis, chromatography systems and others can be reprocessed, but a PDF file or printout is fixed or static and the ability to expand baselines, view the full spectrum, reprocess and interact dynamically with the data set would be lost in the PDF file or printout. As another example, preserving the dynamic format of clinical study data captured in an electronic case report form (eCRF) system allows searching and querying of data, whereas a PDF file of the eCRF data, even if it includes a PDF file of audit trails, would preclude such search and query of the content System Security
— All systems shall be designed and configured with adequate security measures to prevent unauthorized access, changes or deletion of data. Examples are given as below but are not limited to » Individual Login IDs and passwords shall be allotted to users of the system » No shared Login credentials shall be assigned » A list of authorized users with privileges shall be maintained » Administrator access shall be controlled and other users shall not have access to change clock settings and deletion of any data » System administrator should be an independent person who is not involved or interested in outcome of data generated » Physical security shall be provided for servers and PLC nodules — Usage of electronic signatures must have appropriate controls to ensure identity and traceability ƒ Audit Trails
— The purpose of an audit trail for electronic record systems is to provide assurance of the integrity of the electronic record and the associated raw data. Audit trails can be particularly appropriate when users are expected to create, modify, or delete regulated records during normal operations Audit Trail Regulatory Requirements
» Even if there are no predicate rule requirements to document, for example, date, time, or sequence of events in a particular instance, it may nonetheless be important to have audit trails or other physical, logical, or procedural security measures in place to ensure the trustworthiness and reliability of the records. The audit trails shall always be enabled, or other appropriate measures deployed, on the need to comply with predicate rule requirements, a justified and documented risk assessment, and a determination of the potential effect on product quality and safety and record integrity » The system shall enable the recording of the unique identity of operators entering or confirming critical data. Any entry or alteration of critical data shall be authorized and recorded with the reason for the change. Consideration shall be given to building into the system the creation of a complete record of all entries and amendments (a system generated ‘audit trail’). Audit trails need to accurately reflect changes. For example, if a relevant electronic record is created using a number of data fields, all these data fields need to be linked within the audit trail. The aim is to know at any given time point what the information was. Audit trails need to be available and convertible to human readable form — Content of the Audit Trail » The audit trail shall be inextricably linked to the electronic record. It shall be secure and not have the facility for editing or deleting, hence providing a permanent record » The main function of the audit trail is to provide assurance for the integrity of the electronic record. For each entry the following information shall be recorded • Date and time stamp • Name of the user making the change (unique ID) • Link to the record (Batch No, Record ID) • Original value • Changed value • Reason for change » This shall provide the same level of assurance to the record integrity as that of a paper record, that is, if in a process a correction or change is made and the operator makes a correction striking though the initial value, enters the new value, provides reason for change and signs and dates the entry, recording the data and events on paper or through electronic means must have the same level of assurance of data integrity » The audit trail can also provide a record of invalid attempts to log on to the system, to demonstrate the security of the system — Electronic copies can be used as accurate reproductions of paper or electronic records, provided that the copies preserve the content and meaning of the original data, which includes associated metadata, while preserving the static or dynamic nature of the original records — Systems with audit trails » Where systems have an audit trail, the organization shall develop processes for reviewing the audit trail for assuring record integrity. The checking of an audit trail can be labor intensive and therefore the most cost effective method shall be sought. Where records receive a final approval (batch record, change control or deviation), the point of approval of the record may be the most efficient time to check the record. For large systems, such as Enterprise Resource Planning (ERP) systems where thousands of transactions can take place, an audit trail review by exception may be taken, that is, when an error is detected. Personnel responsible for record review under CGMP shall review the audit trails that capture changes to critical data associated with the record as they review the rest of the record » Audit trails that capture changes to critical data shall be reviewed with each record and before final approval of the record. Audit trails subject to regular review shall include, but are not limited to, the following: the change history of finished product test results, changes to sample run sequences, changes to sample identification, and changes to critical process parameters » Processes shall be designed so that quality data required are created, maintained and are prevented from modification. For example, chromatograms shall be sent to long‑term storage (archiving or a permanent record) upon run completion instead of at the end of a day’s run » Storing data electronically in temporary memory, in a manner that allows for manipulation, before creating a permanent record, i.e. electronic data that are automatically saved into temporary memory do not meet CGMP documentation or retention requirements » There shall be technical and procedural controls to meet CGMP documentation practices for electronic systems. For example, a computer system, such as a Laboratory Information Management System (LIMS) or an Electronic Batch Record (EBR) system can be designed to automatically save after each separate entry. This would be similar to recording each entry contemporaneously on a paper batch record to satisfy CGMP requirements. The computer system could be combined with a procedure requiring data be entered immediately when generated » Whatever methods are employed, the approach shall be justified and documented — Systems without audit trails » Where systems do not provide the facility to change data, for example a data logging system used to capture process data, like temperature, and the data is stored securely, an audit trail shall not be necessary. However, such a decision must be documented » In the case where an audit trail is not being employed, a security process must be put in place, validated and controlled via Standard Operating Procedures (SOPs). This shall ensure that only authorized users have access to the system, controls between configuration and operation are separated and that only administrators have access to the operating system » Alternative methods shall be employed to provide assurance of the integrity of critical parameters. Procedures can be put in place to verify, prior to use, the critical parameters (including Control and Alarm Setpoints) to assure that they are set to the validation and process requirements. This shall be documented within the batch record for computerized control systems and in laboratory analysis or sample records for laboratory instrumentation — The organization shall purchase and upgrade software that includes electronic audit trail functionality — Audit trail functionalities should be configured properly to capture general system events and activities relating to the acquisition, deletion, overwriting of and changes to data — Audit trails should be verified during validation of the system — Where audit trail facilities are not available, alternative arrangements must be implemented, e.g. administrative procedures, secondary checks and controls — Audit trail functionality must be enabled and locked at all times — Policy and processes shall be implemented for the review of audit trails in accordance with risk management principles — Audit trails of each batch should be reviewed prior to the release of the batch — Ongoing reviews of audit trails shall be conducted by quality unit based on criticality and complexity of system — Procedure shall be in place to address and investigate any discrepancy found in audit trail ƒ Configuration and design control in IT systems
— The validation activities shall ensure that the configuration settings and design controls for GDP are enabled and managed across the computing environment, including both the software application and operating systems environment. For example, such activities shall include, but are not limited to » Documenting configuration specifications for commercial off‑the–shelf (COTS) systems as well as user–developed systems, as applicable » Restricting security configuration settings for system administrators to independent persons, where technically feasible » Disabling configuration settings for system administrations to independent persons, where technically feasible » Disabling configuration settings that allow over‑writing and reprocessing of the data without traceability » Restricting access to time/date stamps — For systems to be used in clinical trials, configuration and design controls shall be implemented to protect the blinding of the trial, for example, by restricting access to randomization data that may be stored electronically — Data security will be designed so as to protect the data from loss and unauthorized access ƒ Data capture and entry
— Manual entry should be made by authorized individuals only and the system should record details such as who made the entry and when it was made — Data should be entered in specified format which is controlled by the software — All manual entries should be verified by a second operator or by computerized means — Audit trail shall capture changes made — Validation shall be performed of the interface between the data generation, acquisition, processing and recording systems to ensure the accuracy and completeness of data — Data which are captured by the system should be stored in a format that is not susceptible to manipulation or loss — Time stamp shall be generated automatically by the system when data is entered — Procedures shall be in place for any changes and modifications to original data. Changes made must be documented, reviewed and approved ƒ Review of electronic data
— Critical data, identified through risk assessment, shall be reviewed and verified to conclude that activities were executed correctly and changes made to original data, if any, are authorized — The review of data‑related audit trails should be part of the routine data review — SOPs shall be in place that describes how to review audit trails including actions to be taken on identification of any serious issues which may have impact on product quality ƒ Storage, archival and disposal of electronic data
— Data must be stored in its entirety along with metadata, including audit trails — Control shall be put to prevent data storage on unauthorized media such as USB drives, etc. — Written procedures shall govern periodic data archival and backup processes — Backed up data shall be stored in physically isolated locations to survive in the event of disasters and shall be secured to prohibit unauthorized access, alteration and deletion — A procedure for restoration of data shall be available to allow reconstruction of the activity in future — Approved procedures should be in place for the disposal of electronically stored data ƒ Data lifecycle
— Data Lifecycle Process Mapping (DLPM) shall be conducted by the organization. The objective of the process mapping shall be to identify the risk to data integrity in the current process of acquiring, processing, reviewing, reporting and retaining data forms. The outcome of the mapping shall include suggestions for redesigning the data process including data flow and associated business process, as well as listing residual risk and proposed frequency for review and monitoring of this risk to identify opportunities for continuous improvements — Data lifecycle process mapping and associated risk assessment will be done for each business process to identify all data and records generated in each process — DLPM and risk assessment will help the organization identify current gaps in the system and will give the organization the opportunity to implement additional effective controls — Data lifecycle process mapping will be performed with a pre‑defined protocol. Refer to the following Annexures » Annexure 3: Reference Protocol for Data Lifecycle Process Mapping for Manufacturing Stage: Granulation » Annexure 3a: Probability of Errors generated from various Data Generation Sources and Mitigation Plans; and » Annexure 3b: Monitoring of Manufacturing Process and Recording of Process Parameters with their Quality Attributes) — Process mapping shall be done for each process throughout the lifecycle of a drug. The process with the individual documents involved in the process from start point to end point, each process step, description, data involved in the process, SOPs, etc., shall be reviewed against the mapping — In addition to this, the equipment, instruments, and software involved in the process shall be identified. Equipment and instruments including those generating the electronic data shall be identified. Separate data lifecycle process mapping shall be performed for stand‑alone systems — Brain‑storming sessions shall be conducted to evaluate all challenges pertaining to process gaps with SMEs with cross functional training, to address the gaps. Finally, gap closure shall be performed to identify critical priorities and the actions resulting from the same shall be implemented for better compliance with respect to all processes — Validation shall include assessing risk and developing quality risk mitigation strategies for the data lifecycle, including controls to prevent and detect risks throughout the steps of » Data creation and capture » Data processing » Data review » Data reporting, including handling of invalid data and atypical data » Data retention and retrieval — For example, validation activities might include, but shall not be limited to » Determining the risk‑based approach to reviewing electronic data and audit trails based upon process understating and knowledge of potential data impact to product and patient » Writing SOPs defining review of the original electronic records and including meaningful metadata such as audit trails and review of any associated printouts or PDF records » Documenting the system architecture and data flow including flow of electronic data and all associated metadata, from the point of creation through till archival and retrieval » Ensuring that the relationship between data and metadata are maintained intact throughout data lifecycle ƒ SOPs and training
— The validation activities shall ensure that adequate training and procedures are developed prior to release of the system for GXP use. These shall address » Computerized systems administration » Computerized system use » Review of electronic data and meaningful metadata, such as audit trails, including training that may be required in system features that provided users with the ability to efficiently and effectively process data and review electronic data and metadata — Validation shall also cover controls to ensure that good data management practices, for both electronic data and associated paper data, are implemented as deemed appropriate for the system type and its intended use — Data process shall be designed to adequately mitigate and control and continuously review the data integrity risks associated with the steps of acquiring, processing, reviewing, and reporting data, as well as the physical flow of the data and associated metadata across this process through storage and retrieval — Good data process design shall ensure and enhance controls, for each step of the data process, wherever possible, such that each step is » Consistent » Objective, independent and secure » Simple and streamlined » Well‑defined and understood » Automated » Scientifically and statistically sound » Properly documented according to GDP Data collection and reporting: All data collection and reporting will be performed following GDP and applying risk‑based controls to protect and verify critical data ƒ
Data processing: To ensure data integrity, data processing should occur in an objective manner, free from bias, using validated/qualified or verified protocols, processes, methods, systems, equipment and according to approved procedures and training programs ƒ
Data review and data reporting: Data shall be reviewed and, wherever appropriate, evaluated statistically after completion of the process to determine whether outcomes are consistent and compliant with established standards. The evaluation should take into consideration all data, including atypical or suspect data or rejected data, together with the reported data. This shall include a review of the original and electronic records ƒ
Data retention and retrieval: Retention of paper and electronic records has been discussed in the section above, including measures for backup and archival of electronic data and metadata ƒ
Data management of quality processes: Quality processes that have impact on data reliability shall be identified across the manufacturing, packaging, warehousing, engineering, quality‑control and quality assurance operations. All these processes, like recording of batch manufacturing activities, batch testing activities, batch packaging activities, operation of GMP software, handling of QMS activities, document control, validations, reviews, etc. shall be visited for their design so as to evaluate if these are user‑friendly for supporting data reliability. Quality processes shall be inspected for data recording design and shall be improved with the objective of preventing accidental breaches. (Refer to Annexure 5: Data Quality Design Considerations and Controls) ƒ
Exclusion of CGMP data
Any data created as part of a CGMP record must be evaluated by the quality unit as part of release criteria and maintained for CGMP purposes. Electronic data generated to fulfill CGMP requirements shall include relevant metadata. In order to exclude data from the release criteria of the decision‑making process, there must be valid, documented, and scientific justifications for its exclusion. The requirements for record retention and review do not differ depending on the data format; paper‑based and electronic data record‑keeping systems are subject to the same requirements
10. Technology and IT Systems 技术与IT系统Technology and IT systems are helpful in achieving data reliability. Opportunities of uses of IT systems shall be rolled out across the quality related processes ƒ
The organization shall continuously evaluate the IT options that are available and the systems that can be implemented practically. A visionary approach shall be derived and implemented in a timely manner ƒ
The IT framework identifies the elements of IT that shall be considered as a minimum baseline, in managing systems, networks, devices and data, so as to ensure that they are secure, protected appropriately from risk, adequately tested and controlled, and developed and maintained in line with corporate objectives ƒ
The organization shall determine the type of technology that can be implemented based on the complexity, since the amount of controls available in the systems increase with the increase in complexity of the systems. This can be done by identifying risks due to the newly introduced technology, e.g. implementing backup procedure to prevent data loss. Also, the organization shall evaluate the amount of reduction in the initially identified risk by applying the controls that the technology facilitates to mitigate the identified risk ƒ
The organization shall also ensure that the system is selected, implemented and used as per documented procedures, i.e. procedures for qualification and validation of equipment and software. Appropriate installation and operational qualifications should demonstrate the suitability of the computer hardware and software to perform assigned tasks and handle all respective challenges with respect to data integrity ƒ
Validation of Workflow on Computer Systems
— A workflow, such as creation of an electronic Master Production and Control Record (MPCR), is an intended use of a computer system which shall be checked through validation. If the computer system is validated, but it is not validated for its intended use, then it cannot be known for certain whether the workflow will run correctly For example, qualifying the Manufacturing Execution System (MES) platform, a computer system ensures that it meets specifications; however, it does not demonstrate that a given MPCR generated by the MES contains the correct calculations. In this example, validating the workflow ensures that the intended steps, specifications, and calculations in the MPCR are accurate. This is similar to reviewing a paper MPCR and ensuring all supporting procedures are in place before the MPCR is implemented in production ƒ
Data reliability shall be incorporated while evaluating software before implementation ƒ
Vendors shall be evaluated and be required to distribute instruments and equipment, at affordable cost, that will help in global compliance with data integrity requirements ƒ
The following IT SOPs shall be addressed with Technology and IT systems
— IT system maintenance — Physical security — Logical security — Incident and problem management — System change control — Configuration management — Code development — Disaster recovery — Contingency planning — Virus control and systemic software program bug management — Data backup, restoration and retrieval — IT system retirement — Network and server qualification ƒ Following intermediate guidelines shall be addressed with Technology and IT systems
— Overall CSVMP (Computer System Validation Master Plan) — User access control and authority level management — Password lifecycle management — Process and implementation plan of IT systems in quality processes for maintaining data reliability — Electronic signature policy — Risk assessment and mitigation for legacy and stand‑alone systems — Desktop policy — IT administrator policy — Backup policy — Alarm lifecycle management for GXP controls ƒ Development of a robust IT frame work shall include maintaining systems, networks, devices and data, ensuring that they are protected from risk, adequately tested, validated, controlled and maintained ƒ
Electronic signatures with the appropriate controls can be used instead of handwritten signatures or initials in any CGPM required record. An electronic signature with the appropriate controls to securely link the signature with the associated record fulfills this requirement. Electronic signatures should document the controls used to ensure that they are able to identify the specific person who signed the records electronically ƒ
There shall be restriction to alter specifications, process parameters, or manufacturing or testing methods by technical means where possible (for example, by limiting permissions to change settings or data)
ƒOnly authorized personnel shall make changes to computerized MPCRs, or other records, or input laboratory data into computerized records. The organization shall implement documentation controls to ensure actions are attributable to a specific individual ƒ
Login IDs and passwords must not be shared. When login credentials are shared, a unique individual cannot be identified through the login and the system would thus not conform to the CGMP requirements ƒ
The organization shall carry out IT threat assessment like hacking with respect to electronic data, and take a risk‑management approach to protecting data reliability ƒ
All the GMP activities and quality related processes shall be evaluated for implementation of the IT systems with the objective of migrating from paper‑based documentation to electronic documentation, and upgradation of the existing IT systems to improve controls for data reliability requirements. An evaluation and action plan shall be documented, implemented and periodically reviewed ƒ
Electronic systems are categorized as computerized systems and non‑computerized systems ƒ
All computerized systems shall be assessed so as to comply with 21 CFR Part11 /EU annex 11 requirements ƒ
All non‑computerized systems shall be assessed for data integrity risk assessment ƒ
Gaps assessment for legacy IT systems shall be carried out with respect to 21 CFR part 11/EU annexure requirement’s to identify data reliability risk ƒ
Incidents related to computerized systems that could affect the quality of intermediates or APIs or the reliability of records or test results shall be recorded and investigated ƒ
Changes to computerized systems shall be made according to a change procedure and shall be formally authorized, documented, and tested ƒ
Records shall be kept of all changes, including modifications and enhancements to the hardware, software, and any other critical component of the system. These records shall demonstrate that the system is maintained in a validated state ƒ
Risk identification prioritization and mitigation shall be carried out. Appropriate action plan shall be prepared so as to complete the action in a phased manner using procedural controls ƒ
Technology alone cannot entirely eliminate data integrity issues. There are still people and manual processes involved that must be accounted for, monitored, and improved. Therefore, the organization must take a holistic approach to address data integrity issues and apply the necessary designs and controls across all spheres of influence
11. Risk detection and mitigation 风险识别与消除ƒRisk management approach for data reliability implies that risk assessment shall be performed by trained SMEs who will be involved in providing key quality indicators that are affected during data reliability inspection ƒ
Detail risk assessment with respect to data reliability shall be carried out as per ICH Q9 principles. An example related to Data reliability Risk assessment and mitigation evaluation is mentioned in point 5.12 of Annexure-3 Reference Protocol for Data Lifecycle Process Mapping for Manufacturing Stage: Granulation ƒ
All the risks and failures effects associated with the data process lifecycle steps should be identified. There may be more than one failure mode or failure affect for each process step. Once all failure modes have been identified, the scoring can take place to allow ranking of the risk through guidance on quality risk management ƒ
Scores must be provided for failure modes and effects by severity of the risk with data reliability and ease with which these could be detected. The rankings must be justified and rationale should be provided ƒ
Data reliability risk can be classified into three levels: Severity (S), Occurrence (O) and Detection (D). Based on the level of risk classification, overall risk criticality can be identified as high, medium and low ƒ
The risk priority number (RPN) is arrived at by multiplying classification scores, that is, (S) X (O) X (D) ƒ
Potential improvements shall be identified for failures with a RPN. If, however, during evaluation a mitigating activity is identified that can reduce RPN, then this shall be captured as a recommended corrective and preventive action. (Refer to Annexure 7: Risk Assessment Table for Data Recording and Process Control) ƒ
This table is to be used for risk assessment for data recording and process control. Potential improvements shall be identified for failures with an RPN (Risk Priority Number). During evaluation, risk mitigation measures need be identified with current controls that can reduce RPN, and such measures need to be captured as recommended corrective and preventive action. The amount of effort used for risk control should be proportional to the risk, and a suitable due date and classification commensurate with the label of the risk has to be assigned to each section. When it is not possible to reasonably reduce the risk to the acceptable level or to achieve it quickly, a proper justification must be provided ƒ
Risk Management Approach to Data Governance
— Where long‑term measures are identified in order to achieve the desired state of control, interim measures shall be implemented to mitigate risk, and shall be monitored for effectiveness. Where interim measures or risk prioritization are required, residual data integrity risks shall be communicated to senior management, and kept under review. Reverting from automated or computerized to paper‑ based systems will not remove the need for data governance. Such retrograde approaches are likely to increase administrative burden and data risk, and prevent the continuous improvement initiatives — Not all data or processing steps have the same importance to product quality and patient safety. Risk management steps as mentioned in Annexure 3 point 5.12 shall be utilized to determine the importance of each data and processing step. An effective risk management approach to data governance shall take into account data criticality (impact on decision making and product quality). Factors to consider regarding data criticality include
» Which decision/s does the data influence? » What is the impact of the data to product quality or safety? — Risk assessments shall focus on a business process (e.g. production, QC), evaluate data flows and the methods of generating data, and not just consider IT system functionality or complexity. Factors to consider include » Process complexity » Methods of generating, storing and retiring data and their ability to ensure data accuracy, legibility, indelibility » Process consistency and degree of automation / human interaction » Subjectivity of outcome and/or result (i.e. is the process open‑ended or well defined?) 12. Governance 管理Data governance refers to the sum total of arrangements that have been put in place in order to ensure that data, irrespective of the format in which it is generated, is recorded, processed, retained and used to ensure a complete, consistent and accurate record throughout the data lifecycle. The organization shall appoint a Task Force to govern the overall data reliability process. A robust data governance approach will ensure that the data is complete, consistent and accurate, irrespective of the format in which data is generated, used or retained ƒ
Data reliability shall be governed through training and implementation of data reliability related policies including this Guideline ƒ
The organization shall establish and execute an audit program for data reliability that utilizes independent auditors who are qualified by education, experience and training to evaluate the quality systems used for collecting, analyzing, reporting and retaining information and data ƒ
The audit program shall include periodic audit to confirm adherence to established requirements for data reliability ƒ
Management shall be notified of critical data reliability findings that directly impact the quality of products and have potential of serious regulatory concerns. The relevant data reliability information shall be included in the site quality metrics and will be monitored as per a pre‑determined process and frequency ƒ
The data governance structure is provided below
 
Data Governance Structure
Data Reliability Assessment by Data Reliability Auditors
Data reliability assessment shall be part of the self‑inspection program of the organization
— Data Reliability Auditors will be responsible for performing scheduled/unscheduled data reliability assessments (DRA) and inspections at sites as per the authorized data reliability reference checklists referred to below. (Refer to Annexure 4: Data Reliability Inspection Checklist, and Annexure 6: 21 CFR PART 11 Assessment) — The Data Integrity Assessment team shall verify that the document contents comply with and reflect the applicable regulations and the relevant parts of applicable currently authorized product/project regulatory applications (Product Specification Files, Manufacturing and Marketing Authorizations). Documents shall be uniquely identified by suitable means, including version identification where appropriate, with a suitable descriptive title. Constituent pages are unambiguously identified in such a way that completeness is evident, e.g. pagination of the document shall follow the protocol Page 1 of 4, Page 2 of 4, etc. — All forms of documentation and records shall be legible and clear with regard to purpose, function and detail, and all types of GXP documents generated or used shall be properly defined and procedures adhered to — Documentation for records shall provide for accurate and sufficient recording of the various processes and evaluation of observations — Records shall be available for review and audit‑inspection over the lifetime of the product as defined by ICH‑Q10 — The data reliability officers shall also verify the applications that manage access to such records for the purpose of use. These shall be configured so that only authorized persons can access appropriate levels of records, and only approved equipment is used to access records, and that records are not altered during use (e.g. the archiving environment and equipment) — The data reliability officer shall verify that records have been made and retained to demonstrate that in manufacturing, testing, monitoring and essential support processes, the applicable quality principles and regulations have been followed and all the precautions, steps, checks and actions have been taken or made as required, so that products conform with the requirements such that patient safety is assured. There must be controls in place to ensure integrity of data — Entries shall be clear, legible and indelible. If an unstable medium (e.g. heat‑sensitive paper) cannot be avoided or if a document is damaged, then a non‑degrading accurate copy must be retained with the original — Manual entries into records shall be made at the time each action is taken and each result is obtained, i.e. contemporaneously, or as soon as possible thereafter, so that all relevant activities, parameters, results and persons responsible are identified. Completed production records shall be signed and dated by the persons performing the operations — Any alteration, change or correction made to an entry on a record must permit the reading of the original information, and the reason for the change must be clearly evident and recorded where possible. The change(s) must be dated, and signed or logged in an audit trail — Manual recording of data generated during critical operations (in writing or by computer data entry) must be checked by an authorized second operator unless such checks are performed by validated electronic means — Any suspicion that the integrity of data has not been maintained shall be investigated — All persons contributing and the source document(s) used shall be identified in the record. All data recorded for the purpose of supporting quality decisions shall be retained, including the primary ‘raw’ data that are consolidated for presentation in the principal record. The traceability of information flow must be ensured — If a record includes information in the form of codes, then that record must also contain either the means to interpret those codes, or an unambiguous reference to a document that explains the meaning of the codes; further, that document must be retained until the retirement or expiry of the last record in which the reference has been used — The proper functioning of devices and systems for recording data shall be regularly checked and verified by suitable means (e.g. calibration, re‑validation) by an authorized person. Systems used to record critical data/information must be equipped with audit trail capabilities ƒ Risk Management Approach to Data Governance
— Where long‑term measures are identified in order to achieve the desired state of control, interim measures shall be implemented to mitigate risk, and shall be monitored for effectiveness. Where interim measures or risk prioritization are required, residual data integrity risks shall be communicated to senior management, and kept under review. Reverting from automated or computerized to paper‑ based systems will not remove the need for data governance. Such retrograde approaches are likely to increase administrative burden and data risk, and prevent the continuous improvement initiatives — Not all data or processing steps have the same importance to product quality and patient safety. Risk management steps as mentioned in point 10 above shall be utilized to determine the importance of each data and processing step. An effective risk management approach to data governance shall take into account data criticality (impact on decision making and product quality). Factors to consider regarding data criticality include » Which decision/s does the data influence? » What is the impact of the data to product quality or safety? — Risk assessments shall focus on a business process (e.g. production, QC), evaluate data flows and the methods of generating data, and not just consider IT system functionality or complexity. Factors to consider include » Process complexity » Methods of generating, storing and retiring data and their ability to ensure data accuracy, legibility, indelibility » Process consistency and degree of automation / human interaction » Subjectivity of outcome and/or result (i.e. is the process open‑ended or well defined?) The outcome of a comparison between electronic system data and manually recorded events (e.g. apparent discrepancies between analytical reports and raw‑data acquisition times) could be indicative of malpractices ƒ Establishing a Data Governance Structure
For establishing a data governance structure, there should be coordination of people, processes, and technology in order to manage and optimise the use of data as a valued enterprise asset. This will determine the exercise of authority, control, and shared decision‑making (planning, monitoring, and enforcement) over the management of data assets
— The Data Governance Maturity Assessment (DGMA) model shall comprise the following » Open discussion with stakeholders at all levels » One‑on‑one interviews, group workshops, consulting SMEs » Pre‑determined questions to be answered and maturity level determined » Current state maturity to be identified, together with the desired state » It must be ensured that the process is educational and continuous The success of the data governance initiative will be highly dependent on a proper Maturity Assessment. A typical set of steps will comprise the following
Step 1 – Identification and preparation for interviews & workshops
Step 2 – On‑site workshops and interviews with key stakeholders in business and IT
Step 3 – Conducting the Maturity Assessment
Step 4 – Assimilation of findings, analysis, prioritisation of gaps and developing roadmap
Step 5 – Presentation of final Maturity Assessment ƒ
Based on the maturity assessment of data governance in an organization, the appropriate data governance maturity model for the organization can be established ƒ
There are a total of 5 levels for identification of data governance of a site or an organization ƒ
The levels are Level 1 (Initial), Level 2 (Managed), Level 3 (Defined), Level 4 (Quantitatively Managed) and Level 5 (Optimizing), as shown in the diagram below

Based on the data maturity assessment, an organization can evaluate the current status of data governance, associated gaps, and identify the probable risks associated with such gaps. It can then respond with improved processes and reach levels 3 and 4 (where there are defined processes, and data is quantitatively managed at an enterprise level) ƒ
Investigation of Wrongful Act:
— The organization shall establish and follow procedures for conducting an independent, fairly balanced and documented review, and if warranted, an in–depth documented investigation of any alleged falsification, fabrication, or other conduct that raises a question about the integrity of data — Such investigation shall be conducted at the direction of legal counsel to help ensure that documents are properly identified and preserved and that the company receives appropriate advice and legal advice regarding the conduct of such investigation. The investigator(s) shall possess the education, experience, and training to enable the person to conduct data integrity investigation — An independent investigation shall serve to identify potential gaps in systems, processes, procedures and/or practices by individuals or the organization that could raise questions about data integrity. Such investigations shall also serve to assess the legal implications of known or suspected wrongful acts and possible reporting to regulatory authorities — Independent investigations into conduct that raises a question about the integrity of data shall identify all person or persons found to be involved during such investigations, and describe in detail their actions or activities related to the conduct. Such investigation shall also determine the scope of the questionable conduct. For example, the investigation shall determine whether the same or similar conduct or practices may have happened in other instances or could have impacted other data, and if so, the investigation needs to be extended to these events, activities, and practices — Data reliability auditors shall bring the discrepancies identified, if any, to the attention of Corporate Quality for review and shall discuss the same with the site quality group as well — Corporate Quality shall notify the discrepancies to the site Quality Head for investigation and CAPAs ƒ
Data Integrity Failure, Observation, Action Plan and Remediation Process
— If data reliability failure is observed during self or regulatory inspection, then the following three key activities shall be undertaken 1. Comprehensive Evaluation 2. Risk Assessment 3. Remediation and Management Strategy 1. Comprehensive Evaluation » During investigation or post inspection, the investigation shall carry out a comprehensive evaluation of the extent of the inaccuracy of the reported data, in order to arrive at a detailed action plan based on the extent of the deficient documentation practice pertaining to data reliability » The organizational structure and personnel responsibilities shall be examined, specifically of the following areas - Nature of management involvement
- SOPs (Standard Operating Procedures)
» The investigation shall determine actual and factual root cause of the problem, i.e. find out who and what the real source of the problem » A comprehensive evaluation shall include a detailed description of strategies and procedures for finding the scope of the problem. Such an evaluation shall be comprehensive, thorough and complete » The evaluation report shall list the records, application and other documents that have been or will be examined » Scope of evaluation shall include interviewing identified people and personnel involved in the process » The investigation shall examine those involved in the data integrity breach and other related systems that could have the same problems with raw materials, components and ingredients, testing records, production and process records, and equipment 2. Risk Assessment » The organization shall assess the potential effect on the drug product and drug substance quality manufactured and released for distribution to all markets » The organization shall determine the effect of deficient documentation practices on the quality of the drug product released for distribution. Following issues will be evaluated - Were Out–of–specification (OOS) drugs shipped?
- If yes, what is impact on patients?
- Even if no OOS drugs were shipped, it will be important for the organization to maintain appropriate preventive controls
3. Remediation and Management Strategy » Data reliability identification and associated CAPA (Corrective Action and Preventive Action plan) - Management strategy shall include the details of global corrective actions and preventive actions
» The key elements of CAPA will be - Recommendation of third party auditors and inspection
- Identification of responsible persons
- Procedures for monitoring the plan
» Global CAPA for data reliability shall always strive to implement technological controls over procedural controls. The organization shall take advantage of the latest technologies and evolution thereof and move towards a zero‑tolerance approach on data integrity » As part of CAPA, the site where data reliability issue has been observed shall describe the actions the site management responsible are taking or will take, such as contacting customers, recalling product, conducting additional testing and or adding measures to the stability programs to assure stability, monitoring of complaints, and other steps to assure quality of product manufactured under the violative conditions » In addition to CAPA, the site management will also be required to describe other actions, such as revision of procedures, implementation of new controls, training or re‑training of personnel, etc., to prevent recurrence of CGMP violations, including breaches of data reliability, using the data governance plan discussed earlier in this Guideline » The organization shall take corrective action as required to confirm the accuracy, completeness and truthfulness of all the data and information contained in the submissions(s) and provide the regulatory authority with corrected or additional data or information as applicable ƒ Data Reliability Remediation
— Remedial measures in relation to data reliability shall be designed with the objective of reconstructing the process through actual available records and reports, with the complete negation of any possibility of data falsification, omission, hiding and substitution — While carrying out remediation, a comprehensive remediation plan shall be prepared by the organization in order to carry out investigation based on actual and factual data to understand the probable root cause(s) of the problems, e.g. lack of awareness, intentional act, emphasis on quantity over quality, shortage of manpower, performance and business pressure, inadequate process and technology, lack of effectiveness of training or any other reason(s). Accordingly, the organization shall prepare the appropriate Corrective and Preventive Action (CAPA). There shall be full Management support to bring and enhance quality culture elements for building better data reliability culture — The organization shall be committed to voluntary remediation activities along with regulatory bodies in order to correct the problems through detailed examination of inefficient processes — Management approach and monitoring shall be driven by a zero‑tolerance approach to data integrity 13. References 参考资料 |
评分
-
查看全部评分
|


 |手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033
|手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033





 发表于 2017-3-31 16:29:04
发表于 2017-3-31 16:29:04
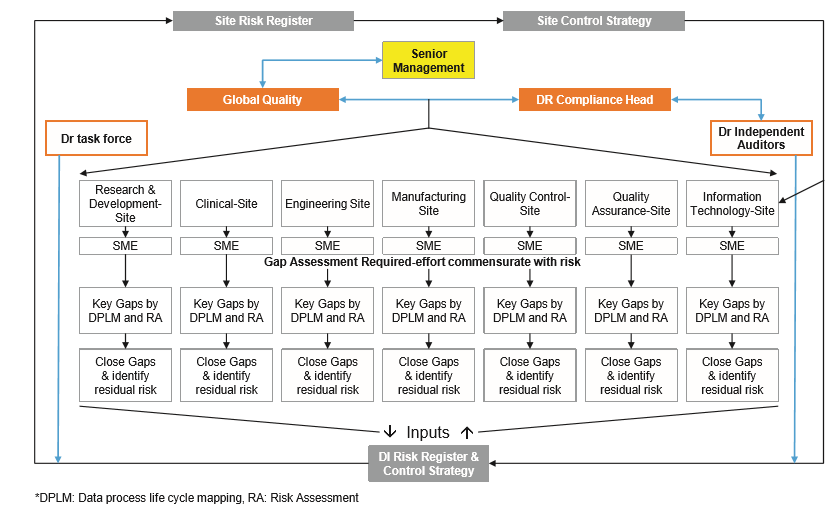
 置顶卡
置顶卡 变色卡
变色卡 楼主
楼主
 可以发个网盘链接
可以发个网盘链接



